

Polyvinylpyrrolidone PVP coprecipitate
At present, about 40% of the drugs used on the market are insoluble drugs, because they are difficult to absorb and utilize in the human body, and the use of drugs is limited to a certain extent. In order to increase the dissolution and absorption of insoluble drugs, researchers have developed many methods from various aspects of pharmaceutical and pharmaceutical, such as drug salting, micropulverization, cyclodextrin inclusion, preparation of amorphous, Solid dispersion, and Co-crystal (co-precipitation Co-precipitate). Among the preparation of drug co-precipitation, drug-polyvinylpyrrolidone co-precipitation is one of the most studied. The principle, significance and method of preparation of drug-polyvinylpyrrolidone (PVP) co-precipitate are reviewed in this paper.
1. Introduction to polyvinylpyrrolidone
Polyvinylpyrrolidone (Povidone, PVP) is a synthetic polymer with a chain structure of vinyl. The structural formula (C6H9NO)n is shown as follows:
Figure 1. Chemical structure of polyvinylpyrrolidone
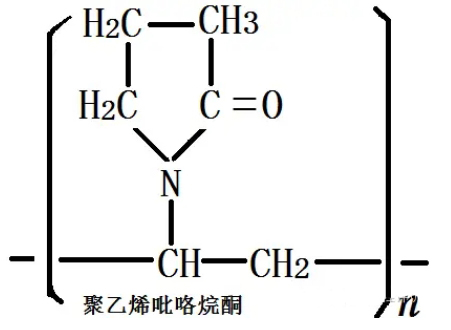

Since PVP is a polymer, the drug enters the PVP skeleton in the form of a single molecule after co-precipitation with PVP. When PVP is dissolved in water, the drug can be released, eliminating the dissolution process. This is why drug-PVP coprecipitates can improve the solubility of insoluble drugs.
PVP is a white, odorless, tasteless powder, easy to absorb moisture, dissolved in water, ethanol, chloroform, isopropyl alcohol, insoluble in acetone and ether.
This product can be used as a co-solvent for tablets, granules and injections, as a flow aid for capsules, as a dispersant for liquid preparations and colorants, as a stabilizer for enzymes and heat-sensitive drugs, as a co-precipitation agent for insoluble drugs, as a detoxifying agent for ophthalmic drugs and as a lubricant.
2. Mechanism and significance of preparation of drug-PVP coprecipitate
Many studies have proved that the co-precipitate of insoluble drugs and PVP can significantly change the solubility of insoluble drugs, and thus achieve the purpose of increasing absorption and bioavailability. Some drugs have good efficacy, but their fatal disadvantage is that the solubility in water is very small, resulting in their bioavailability is greatly reduced, the use of some water-soluble substances with these drugs co-precipitation, and then improve the solubility and dissolution rate of drugs, to reduce the dose, improve the effect of curative effect. As a co-precipitant of insoluble drugs, PVP is being widely used.
As for the mechanism of action of co-precipitates, most people believe that drugs and carriers such as PVP in the solvent evaporation process, due to hydrogen bonding, recombination and viscosity increase, the carrier inhibits the formation of drug nucleation and crystal growth, so that the drug becomes an amorphous substance with higher energy, thereby improving the saturation of the drug. Some people think that PVP forms a network on drug molecules or drug molecules enter the mesh skeleton of PVP molecules, preventing the formation of drug crystal structure.
The main reason why PVP is used as a drug coprecipitator is that carbonyl O in PVP molecules can be combined with active hydrogen bonds in insoluble drug molecules, which on the one hand makes relatively small drug molecules enter into PVP macromolecules in an amorphous state; on the other hand, hydrogen bonds do not change the water-soluble nature of PVP. As a result, insoluble drug molecules are dispersed in PVP macromolecules by hydrogen bonds, making them easily soluble.
Table 1 Solubility changes of some insoluble drugs after forming coprecipitates with PVP
|
The name of the drug |
Proportion of co-precipitate with PVP |
Solubility is increased by a factor of two |
|
Phenytoin |
1:5 |
2.3 |
|
Amorpholine |
1:5 |
38 |
|
Reserpine (297-420um). |
1:3 |
15 |
The increase of solubility of insoluble drugs in human body also correspondingly improves the bioavailability of drugs. For example, the bioavailability of drugs increased by 1.55 times after phenytoin co-precipitation with PVP, and the increase of solubility after co-precipitation of insoluble drugs was related to the molecular weight of PVP and the amount of PVP. When the amount (quality) of PVP is the same, the increase rate of drug solubility decreases in the order of PVP-K15 > PVP-K30 > PVP-K90, because the solubification effect of PVP itself changes in the order of PVP-K15 > PVP-K30 > PVP-K90, and PVP-K15 is generally used more.
The solubility increase of insoluble drugs and PVP coprecipitates varies with the amount of PVP in a complex way. For PVP with a certain molecular weight, the number of molecules of each PVP molecule can bind to is certain, and insoluble drugs often have a certain crystal state. When the amount of PVP is not enough to bind a certain amount of drugs and make them in an amorphous and dispersed state, The drug was still mainly in crystalline state, and the solubility changed little. When PVP must reach a certain content, the drug appears as an amorphous dispersion system, and its solubility can be significantly increased to achieve the purpose of rapid dissolution and absorption. For different drugs, the amorphous dispersed PVP content is not the same as that of PVP co-precipitation, such as 70% of cyclohexamine acetate, and B-carotene, chloramphenicol, dexamethasone, hydroprednisone, streptomycin, tetracycline and testosterone can be increased in human solubility and bioavailability by PVP co-precipitation.
Bates et al. pointed out that when the ratio of reserpine-PVP coprecipitates was 1:3 and 1:6, the time required for 50% dissolution was 7 minutes and 0.5 minutes, respectively, while the mechanical mixture and pure drug were 35 minutes and 106 minutes, respectively. From the dissolution data, it can be seen that the dissolution rates of 1:3 and 1:6 coprecipitates are 5 and 70 times higher than their mechanical mixtures. More than 15 times and 200 times higher than pure drugs.
Corrigan et al. confirmed that hydrofluoromethyl azide and PVP co-precipitate form amorphous substances and water-soluble complexes due to the inhibition of drug crystallization by PVP, thus increasing the apparent solubility and dissolution rate of drugs. When the concentration of PVP is high, its dissolution rate is 16 times higher than that of pure medicine. At the same time, there was a linear relationship between the apparent solubility and PVP concentration.
3. Preparation of drug-pvp co-precipitation
There are many preparation methods for drug PVP co-precipitation, but the basic method is to use organic solvent to slowly evaporate solvent under normal pressure or vacuum condition to obtain co-precipitation. Common solvents are ethanol, chloroform, dichloromethane, dimethyl sulfoxide, etc. The ratio of drugs to PVP also varies from 1/1 to 1/10.
Li Maoxing et al. obtained the drug PVP coprecipitate by evaporating the solvent with anhydrous ethanol. Accurately weigh the appropriate amount of phenobarbital, a total of 3 parts, respectively on the evaporation dish, with a small amount of anhydrous ethanol heated to dissolve, add PVP according to the weight ratio, stir to completely dissolve, put in 95℃ water bath to steam the solvent, obtain a white glass, dry in a dryer for 24h. Crushed and sifted (60 mesh) to obtain l:3, 1:6, 1:10 phenobarbital -PVP co-precipitate.
Wang Zhong et al. used chloroform as solvent to prepare lacidipine-pvp coprecipitation by drying under reduced pressure. The details are as follows: Weigh a certain amount of PVP and add an appropriate amount of chloroform to dissolve in a water bath of 50°C, add 1 chloroform solution to mix well, and put it in a vacuum drying oven to dry under pressure at 40-50°C for 2h. The coprecipitated is vitreous foam. It was crushed, screened 80 mesh and dried at 50°C for 1h under atmospheric pressure.
Wang Yang used dimethyl sulfoxide as solvent to obtain norfloxacin-PVP co-precipitate and prepared the co-precipitate by neutralization method. Solvent method: Weigh norfloxacin and PVP according to different weight ratios, dissolve in DSO and stir until the clarified solution, slightly heat dissipate the solvent, dry the product in a vacuum dryer, grind through 80 mesh sieve, and put it in a dryer for use. Neutralization method: After weighing norfloxacin and PVP according to different weight ratios, dissolve in 1mol/L NaOH solution, stir until clarified, quickly add 1mol/L HCl solution, quickly stir to make completely neutralized and neutral, and the obtained product is dried in a vacuum dryer.
After the formation of drug-PVP coprecipitates, identification is needed. More commonly used methods are XRD, as well as DSC, DTA technology. When the drug forms co-precipitation with PVP, the crystalline drug will be transformed into amorphous, which can be confirmed by XRD, DSC, DTA and other technologies.
MINORU YOSHIOK et al. prepared indomethin-PVP coprecipitates and identified them by DSC.
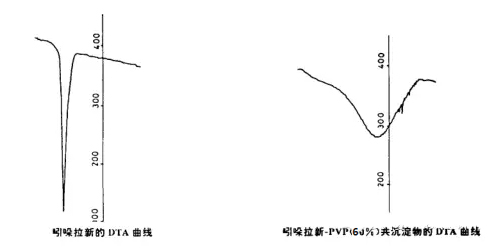
Indolacine-pvp coprecipitates were prepared and identified by DTA. In the left picture of DTA of the original drug indolacine, a sharp endothermic peak appeared near 168~170℃, but a very dull endothermic peak appeared on the DTA curve of the drug-PVP coprecipitation, and the corresponding melting point could not be found, as shown in the right picture of Figure 2, indicating that the drug crystal form in the coprecipitation almost became amorphous and dispersed in PVP.
Figure 2: DTA curves of indoclacine and indoclasin-PVP (60%) co-precipitate
4. Conclusion
The cause of solubility of insoluble drugs can be improved by the formation of co-precipitation through drug PVP. The solubility and dissolution rate of the drug can be improved to reduce the dose and improve the curative effect. As a co-precipitant of insoluble drugs, PVP is being widely used. The development and utilization of co-precipitation technology can improve the quality of drugs and meet the needs of patients for safe and effective drug use. At present, coprecipitation technology remains more in the scientific research stage, and how to apply this technology to production is also a problem that needs to be solved in the future.